CGCNN (Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties)¶
Before starting training and evaluation, please download the dataset and split it. Data reading requires additional dependency pymatgen, please run the installation command pip install pymatgen.
| Pretrained Model | Metrics |
|---|---|
| cgcnn_pretrained.pdparams | loss(MAE): 0.4195 |
1. Background Introduction¶
Machine learning methods are becoming increasingly popular for accelerating new material design, predicting material properties with accuracy close to ab initio calculations but orders of magnitude faster. The arbitrary size of crystal systems poses a challenge because they need to be represented as fixed-length vectors to be compatible with most algorithms. This problem is usually solved by manually constructing fixed-length feature vectors using simple material properties or designing symmetry-invariant transformations of atomic coordinates. However, the former requires individual design to predict different properties, while the latter makes the model difficult to interpret due to complex transformations. CGCNN is a generalized crystal graph convolutional neural network framework for representing periodic crystal systems, which provides both material property prediction with density functional theory (DFT) accuracy and atomic-level chemical insights. Therefore, this case uses CGCNN to predict the band properties of 2D semiconductor materials.
2. Model Principle¶
This chapter only briefly introduces the model principle of CGCNN. For detailed theoretical derivation, please read Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties.
CGCNN is a general machine learning framework for representing periodic crystal systems. Unlike traditional methods that rely on manually constructed feature vectors, CGCNN builds convolutional neural networks directly on top of the Crystal Graph, thereby automatically learning representations to predict material properties with Density Functional Theory (DFT) accuracy and providing atomic-level chemical insights.
Crystal Graph Representation: The crystal structure is converted into an Undirected Multigraph \(G\). * Nodes (\(i\)): Represent atoms. Each node is described by a feature vector \(v_i\), encoding atomic properties (such as group number, period number, electronegativity, etc.). * Edges (\((i,j)_k\)): Represent chemical bond connections between atoms. Due to the periodicity of crystals, there may be multiple edges between the same pair of atoms (multigraph). Each edge is defined by a feature vector \(u_{(i,j)_k}\) corresponding to the \(k\)-th bond connecting atoms \(i\) and \(j\). * Construction method: Usually search for nearest neighbors within a 6 Å radius. If atoms share a Voronoi face and are close enough (based on covalent bond length), they are considered connected.
Convolutional Layers: The core "learning" process occurs in the convolutional layers. The model iteratively updates the feature vector of each atom by aggregating information from surrounding atoms and bonds to capture the local chemical environment. Convolution function: To distinguish the difference in interaction strength between neighbors, the model uses an improved update rule: $\(v_{i}^{(t+1)} = v_{i}^{(t)} + \sum_{j,k} \sigma(z_{(i,j)_{k}}^{(t)} W_{f}^{(t)} + b_{f}^{(t)}) \odot g(z_{(i,j)_{k}}^{(t)} W_{s}^{(t)} + b_{s}^{(t)})\)$ Where: * Concatenation (\(z\)): \(z_{(i,j)_{k}}^{(t)} = v_{i}^{(t)} \oplus v_{j}^{(t)} \oplus u_{(i,j)_{k}}\) is the concatenation of the central atom vector, neighbor atom vector, and bond vector. * Gating (\(\sigma\)): The Sigmoid function \(\sigma(\cdot)\) acts as a learned weight matrix (i.e., gating mechanism), used to automatically distinguish the strength of interactions between different neighbors (e.g., automatically ignoring weak bonds). * Nonlinearity (\(g\)): The function \(g(\cdot)\) adds nonlinear coupling. * Residual connection: Adding the original \(v_{i}^{(t)}\) in the formula makes it easier to train deeper networks.
Pooling and Output: After \(R\) convolutional layers, the model needs to generate a fixed-length vector representing the entire crystal structure, regardless of how many atoms are in the unit cell. * Pooling Layer: Uses Normalized Summation as the pooling function. $\(v_{c} = \frac{1}{N} \sum_{i} v_{i}^{(R)}\)$ This ensures that the representation has Permutational Invariance of atomic indices and Size Invariance of the unit cell.
- Output Layer: The crystal feature vector \(v_c\) passes through fully connected hidden layers (\(L_1, L_2\)) to capture complex mapping relationships, and finally predicts the target property \(\hat{y}\) (e.g., formation energy, band gap) through the output layer.
The overall structure of the model is shown in the figure:
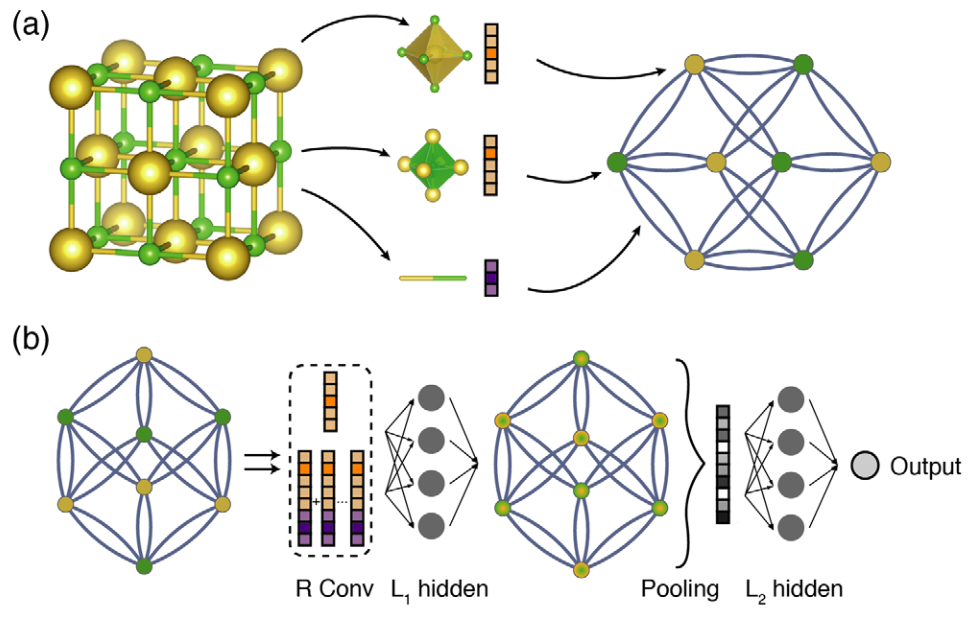
The CGCNN paper predicts seven different properties. Next, we will introduce how to use PaddleScience code to implement the CGCNN network to predict the gap properties of 2D semiconductors.
3.1 Dataset Introduction¶
The original CGCNN paper uses the dataset (https://next-gen.materialsproject.org/) and the dataset (https://cmr.fysik.dtu.dk/cubic_perovskites/cubic_perovskites.html).
The Materials Project dataset is a large-scale open online material database established by the University of California, Berkeley in cooperation with Lawrence Berkeley National Laboratory, dedicated to providing comprehensive material performance data, structural information, and calculation simulation results. The dataset contains data on more than one million inorganic materials from high-throughput first-principles calculations. It includes detailed information such as crystal structure, energy characteristics, electronic structure, thermodynamic properties, providing researchers with rich material data resources. The MPDataDoc object contains a total of 69 fields, of which 57 fields describe the properties of materials from the aspects of material representation, photoelectric properties, mechanical properties (elastic properties, shear properties), physical and chemical properties (chemical composition, physical structure, microstructure), stability and reactivity (also belonging to chemical properties), thermodynamic properties, magnetic properties, etc.
This case uses a self-collected dataset for training and testing. If users need to use this case for related tasks, they can refer to the following dataset format:
- CIF A file used to record the crystal structure required by the user.
- [id _ prop.csv] The target property of each crystal.
You can create a custom dataset by creating a directory root_dir containing the following files:
-
id_prop.csv: CSV The first column re-encodes a uniqueIDfor each crystal, and the second column re-encodes the value of the target property. -
atom_init.json: JSON Stores the initial vector of each element. -
ID.cif: CIF A file that re-encodes the crystal structure, whereIDis the unique ID of the crystal in the dataset.
The structure of root_dir should be (root_dir generally refers to the training/evaluation/test data folder):
3.2 Model Construction¶
CGCNN needs to construct a model through the data used, so CGCNNDataset needs to be instantiated first. After instantiating CGCNNDataset, information such as the length of training samples and input dimensions can be obtained. Based on this information and the set model hyperparameters cfg.MODEL.atom_fea_len, cfg.MODEL.n_conv, cfg.MODEL.h_fea_len, cfg.MODEL.n_h, the instantiation of CrystalGraphConvNet is completed.
The hyperparameters cfg.MODEL.atom_fea_len, cfg.MODEL.n_conv, cfg.MODEL.h_fea_len, cfg.MODEL.n_h are set by default as follows:
| examples/cgcnn/conf/CGCNN.yaml | |
|---|---|
3.3 Constraint Construction¶
The model of this problem is a regression model, trained using supervised learning, so the PaddleScience built-in supervised constraint SupervisedConstraint can be used to construct supervised constraints. The code is as follows:
Where root_dir is the training set path, and batch_size is the batch training size. In order to perform normal batch training, collate_fn needs to be redesigned according to the model. The code for collate_pool is as follows:
3.4 Validator Construction¶
In order to monitor the training status of the model in real time, we will evaluate the model after each round of training. Consistent with the training process, we use the SupervisedValidator function built into PaddleScience to construct a supervised data validator. The specific code is as follows:
3.5 Optimizer Construction¶
The SGD optimizer is used for training. The relevant code is as follows:
| examples/cgcnn/CGCNN.py | |
|---|---|
The training hyperparameters cfg.TRAIN.lr, cfg.TRAIN.momentum, cfg.TRAIN.weight_decay, etc. are set by default as follows:
3.6 Model Training¶
Since this problem is modeled as a regression problem, PaddleScience's built-in psci.loss.MAELoss('mean') can be used as the loss function for the training process. At the same time, stochastic gradient descent is chosen to optimize the network. And the training process is encapsulated in the Solver built into PaddleScience. The specific code is as follows:
| examples/cgcnn/CGCNN.py | |
|---|---|
4. Complete Code¶
| examples/cgcnn/CGCNN.py | |
|---|---|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 | |