Before starting training and evaluation, please download the data file data_set.xlsx, and modify data_dir in the yaml configuration file to the actual file path of data_set.xlsx.
If you need to use a pre-trained model for evaluation, please download the pre-trained model smc_reac_model.pdparams, and modify load_model_path in the yaml configuration file to the model parameter path.
Before the first training and evaluation, please execute pip install -r requirements.txt to install rdkit and other related dependencies.
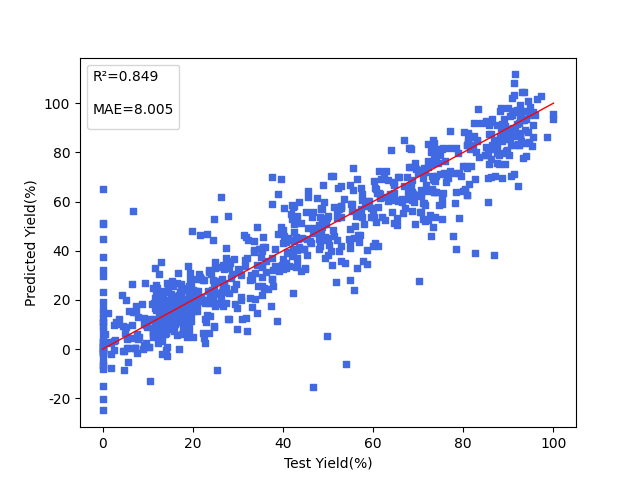
Catalyzed by zero-valent palladium complexes, aryl or alkenyl boronic acids or boronic esters undergo cross-coupling with chloro-, bromo-, iodoarenes or alkenes. This reaction has the advantages of mild reaction conditions and high conversion rate, and plays an important role in fields such as material synthesis and drug development, but has problems such as long development cycle and high trial and error cost. This study establishes a prediction model by using high-throughput experimental data to analyze the effects of reaction substrates (including electrophiles and nucleophiles), catalytic ligands, bases, and solvents on the yield of coupling reactions.
2. Implementation of Suzuki-Miyaura Cross-Coupling Reaction Yield Prediction Model¶
This section will explain how to implement the construction, training, testing and evaluation of the Suzuki-Miyaura cross-coupling reaction yield prediction model based on PaddleScience code. The directory structure of the case is as follows.
The data used in this example comes from the open source data provided in reference [1], considering only the influence of reagents themselves on experimental results, and filtering out partial reaction data where reagents participated in all components, saved in the file ./data_set.xlsx. This work developed an automated platform based on flow chemistry, which was assembled in an argon-protected glove box, using a modified high-performance liquid chromatography (HPLC) system combined with an automated sampling device to draw reaction components (electrophiles, nucleophiles, catalysts, ligands, and bases) from 192 reservoirs according to a set program and inject them into the flow carrier liquid. Each reaction segment reacts in a temperature-controlled reaction coil at a set flow rate, pressure, and time, and the reaction solution is detected in real time by UPLC-MS. By regulating the combination of electrophiles, nucleophiles, 11 ligands, 7 bases, and 4 solvents, a systematic screening of 5760 reaction conditions was finally achieved. Next, taking one piece of data as an example, combined with code to illustrate the construction and loading process of the dataset.
Apply rdkit.Chem.rdFingerprintGenerator to convert the SMILES descriptions of electrophiles, nucleophiles, catalytic ligands, bases, and solvents into Morgan fingerprints. Morgan fingerprint is a vectorized description of molecular structure, encoded as hash values through local topology, and mapped to 2048-bit fingerprint bits. Expressed in PaddleScience code as follows
This case uses supervised learning. According to the PaddleScience API structure description, the built-in SupervisedConstraint is used to construct supervised constraints. Expressed in PaddleScience code as follows
The second parameter of SupervisedConstraint indicates using mean squared error MSELoss as the loss function, and the third parameter indicates the name of the constraint condition, which is convenient for subsequent indexing.
This case designed five independent sub-networks (fully connected layer + ReLU activation), each sub-network extracts features of corresponding chemical substances respectively. Subsequently, these five feature vectors are weighted averaged through trainable weight parameters to achieve adaptive learning of the impact of different chemical components on reaction yield prediction. Finally, the fused features are input into a fully connected layer for further mapping to output the predicted value of reaction yield. The entire network structure reflects the independent extraction and weighted fusion of information of each component in the reaction, consistent with the characteristics of the reaction mechanism. Expressed in PaddleScience code as follows
After completing the above settings, you only need to pass the instantiated objects to ppsci.solver.Solver in order, and then start training. Expressed in PaddleScience code as follows